Curation and Analysis of a Saccharomyces cerevisiae Genome-Scale Metabolic Model for Predicting Production of Sensory Impact Molecules under Enological Conditions


Abstract
:1. Introduction
2. Materials and Methods
2.1. Model Curation and Development
2.2. Flux Balance Analysis
2.3. Flux Variability Analysis
2.4. Computing Environment
3. Results
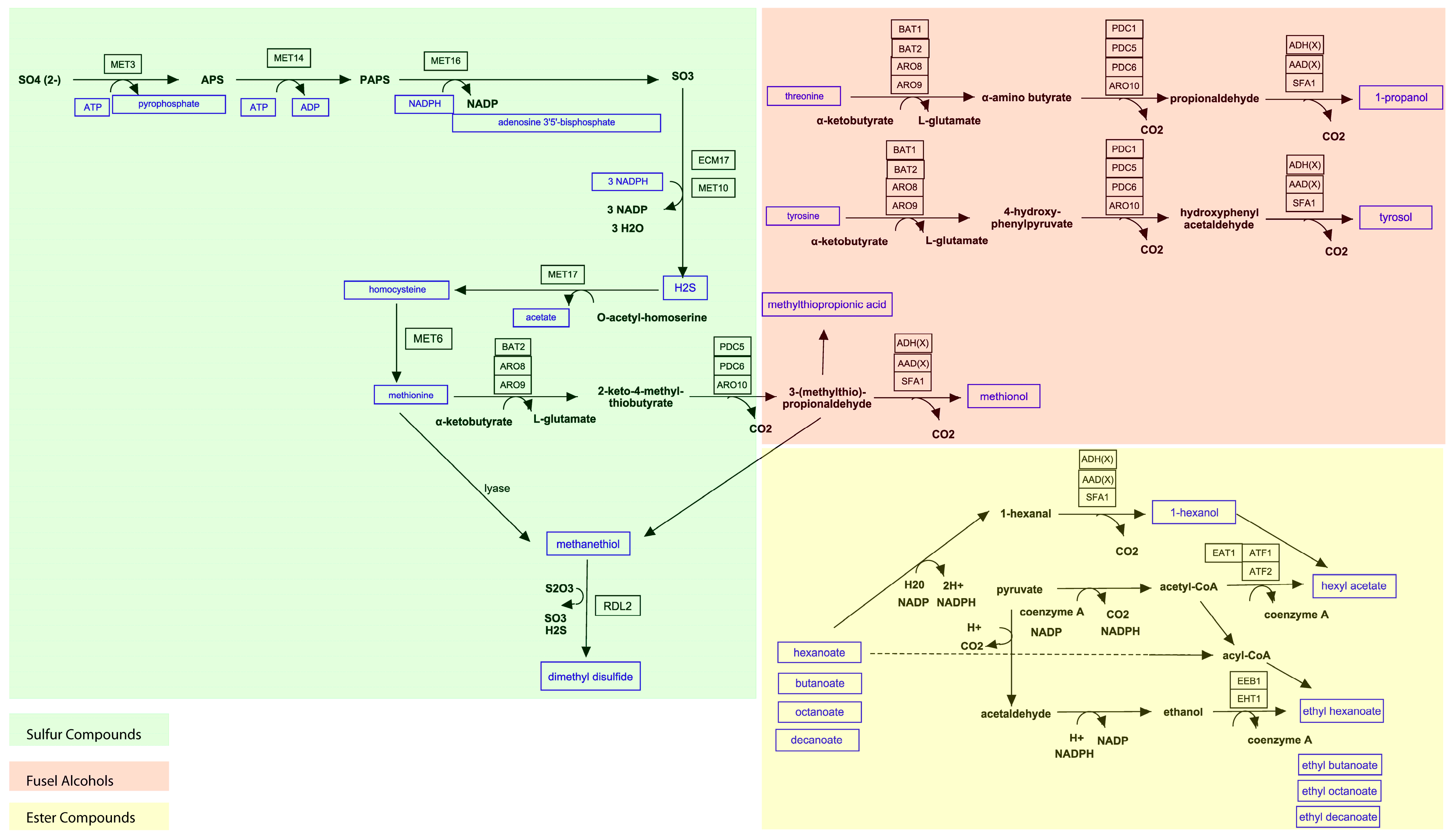
3.1. Coverage of Aroma Pathways in the Model (GSMM)
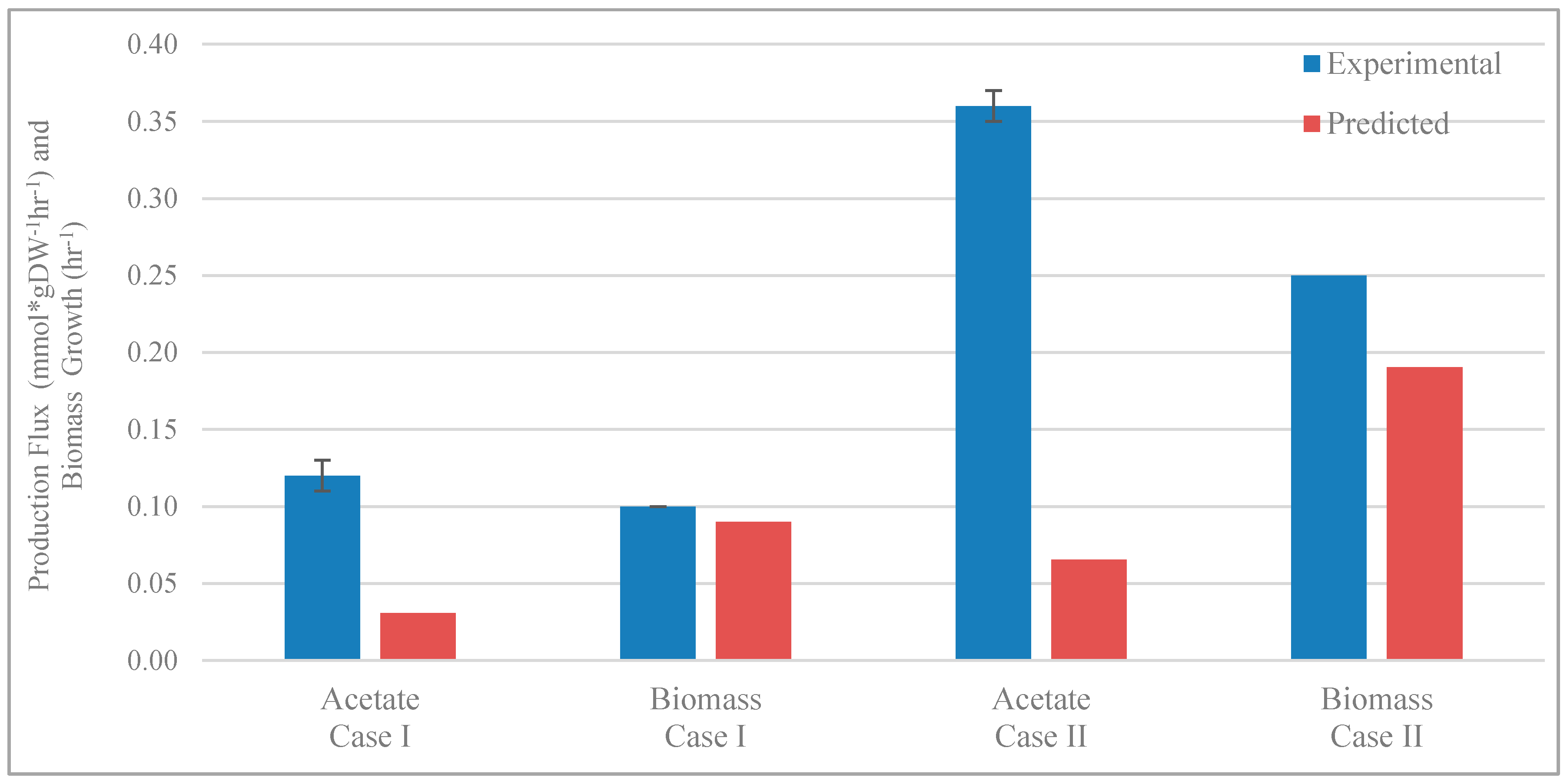
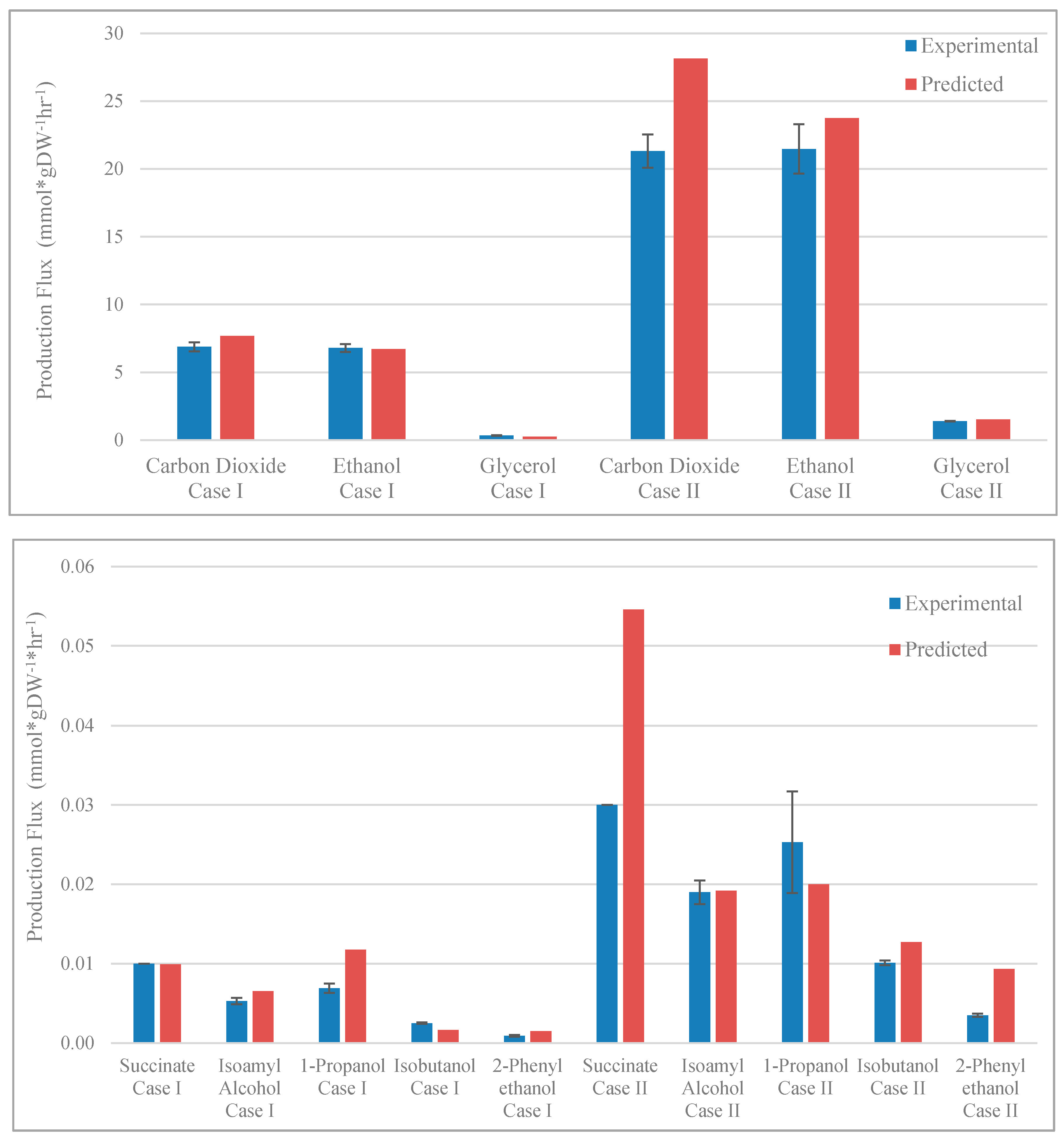
3.2. Validation of Model Predictions
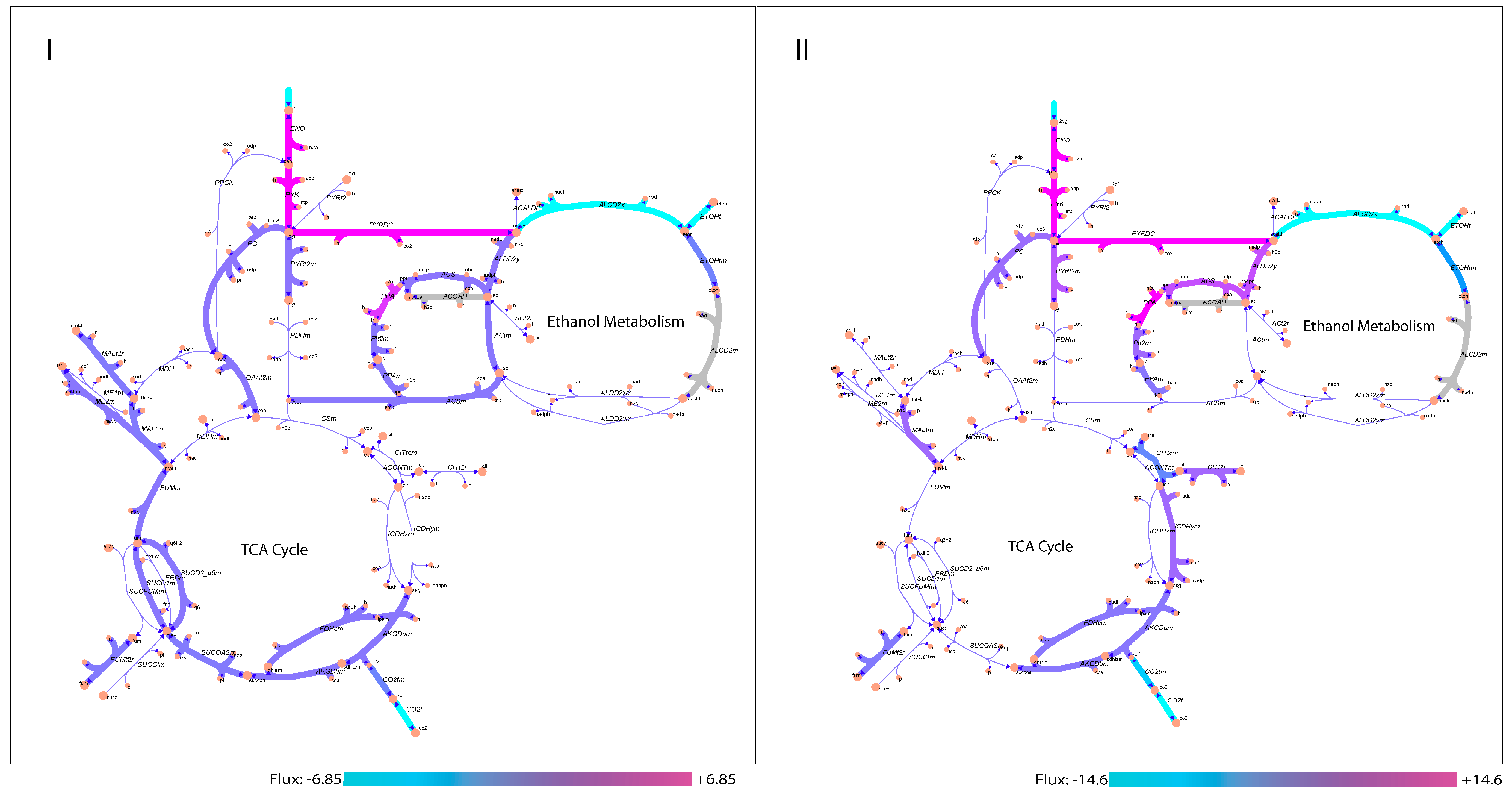
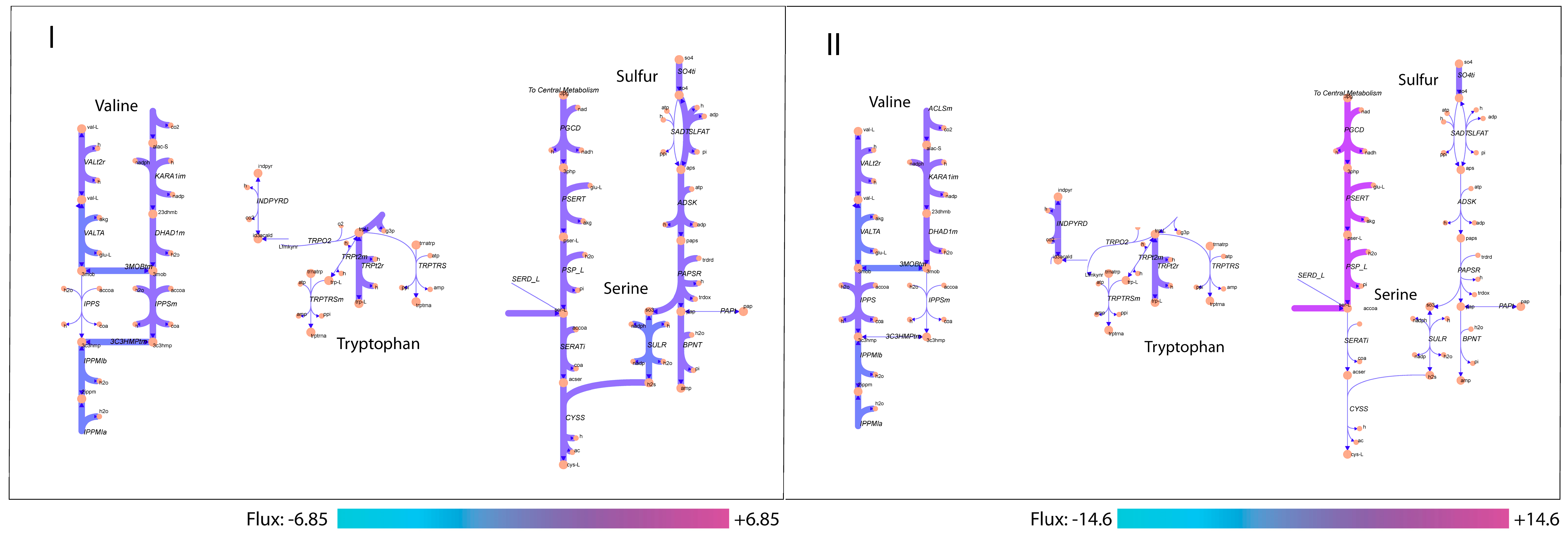
3.3. Analysis of Active Aroma Reactions in Metabolic Network
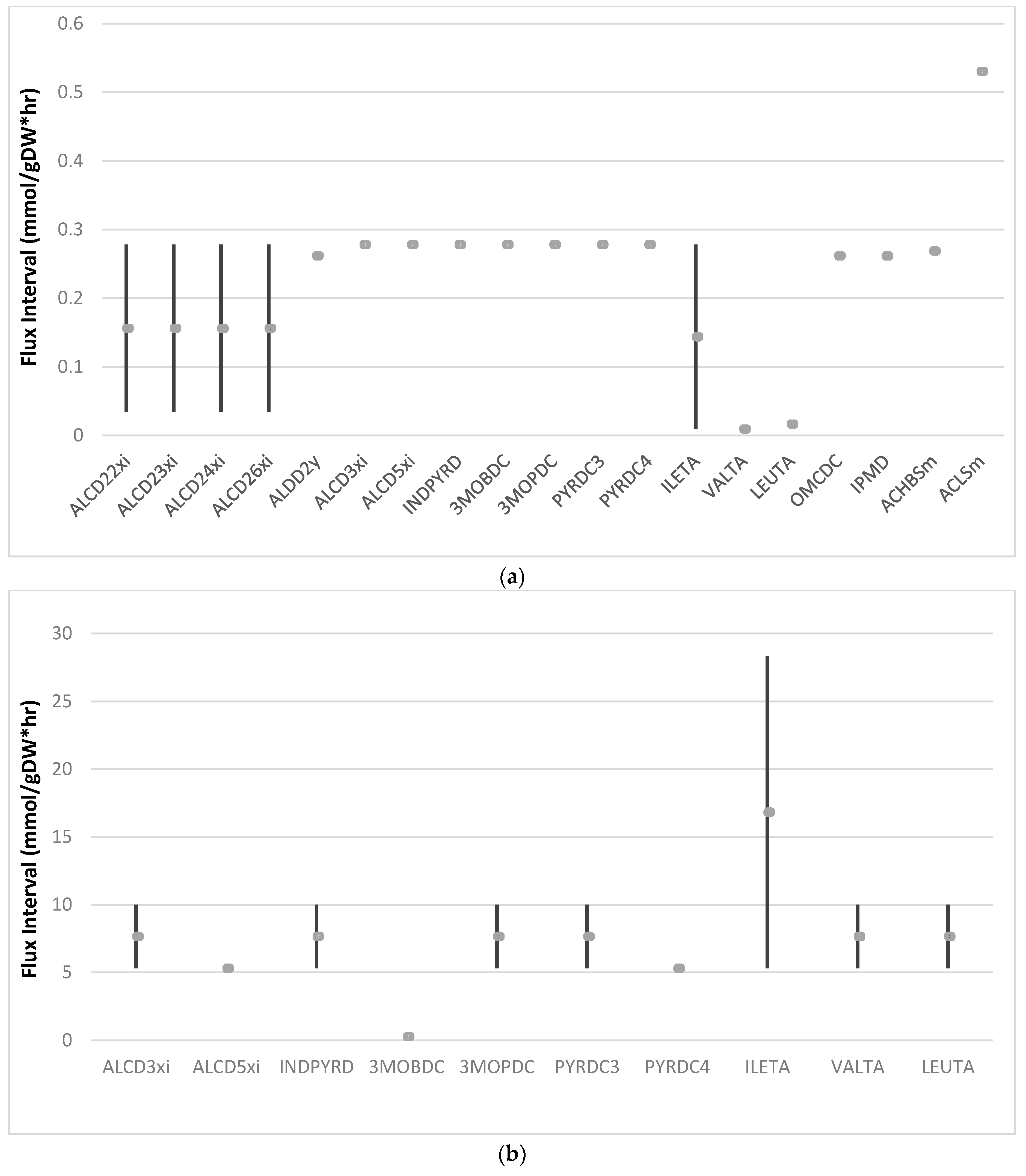
3.4. Exploring Network Flexibility via FVA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FBA | Flux Balance Analysis |
| FVA | Flux Variability Analysis |
| GSMM | Genome-scale metabolic model |
| MCFA | medium-chain fatty acid |
| ALCD22xi | aldehyde dehydrogenase (via 2 methylbutanol NAD) |
| ALCD23xi | aldehyde dehydrogenase (via isobutyl alcohol NAD) |
| ALCD24xi | aldehyde dehydrogenase (via isoamyl alcohol NAD) |
| ALCD26xi | aldehyde dehydrogenase (via tryptophol NAD) |
| ALDD2y | aldehyde dehydrogenase (via acetaldehyde NADP) |
| ALCD3xi | aldehyde dehydrogenase (via propanol NAD) |
| ALCD5xi | aldehyde dehydrogenase (via tyrosol NAD) |
| INDPYRD | indole-3 pyruvate decarboxylase |
| 3MOBDC | 3 methyl 2 oxobutanoate decarboxylase |
| 3MOPDC | 3 methyl 2 oxopentanoate decarboxylase |
| PYRDC3 | pyruvate decarboxylase (aldehyde forming) |
| PYRDC4 | pyruvate decarboxylase (hydroxyl phenyl) |
| ILETA | isoleucine transaminase |
| VALTA | valine transaminase |
| LEUTA | leucine transaminase |
| OMCDC | 2 oxo 4 methyl 3 carboxypentanoate decarboxylation |
| IPMD | 3 isopropylmalate dehydrogenase |
| ACHBSm | 2 aceto 2 hydroxybutanoate synthase (mitochondrial) |
| ACLSm | acetolactate synthase (mitochondrial) |
References
- Vandamme, E.J.; Soetaert, W. Bioflavours and fragrances via fermentation and biocatalysis. J. Chem. Technol. Biotechnol. 2002, 77, 1323–1332. [Google Scholar] [CrossRef]
- Rossouw, D.; Bauer, F.F. Comparing the transcriptomes of wine yeast strains: Toward understanding the interaction between environment and transcriptome during fermentation. Appl. Microbiol. Biotechnol. 2009, 84, 937–954. [Google Scholar] [CrossRef]
- Rossouw, D.; van den Dool, A.H.; Jacobson, D.; Bauer, F.F. Comparative transcriptomic and proteomic profiling of industrial wine yeast strains. Appl. Environ. Microbiol. 2010, 76, 3911–3923. [Google Scholar] [CrossRef] [Green Version]
- Hazelwood, L.A.; Daran, J.M.; van Maris, A.J.A.; Pronk, J.T.; Dickinson, J.R. The Ehrlich pathway for fusel alcohol production: A century of research on Saccharomyces cerevisiae metabolism. Appl. Environ. Microbiol. 2008, 74, 2259–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiegers, J.H.; Pretorius, I.S. Yeast modulation of wine flavor. In Advances in Applied Microbiology; Laskin, A.I., Bennett, J.W., Gadd, G.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 57, pp. 131–175. [Google Scholar]
- Verstrepen, K.J.; Van Laere, S.D.M.; Vanderhaegen, B.M.P.; Derdelinckx, G.; Dufour, J.P.; Pretorius, I.S.; Winderickx, J.; Thevelein, J.M.; Delvaux, F.R. Expression levels of the yeast alcohol acetyltransferase genes atf1, lg-atf1, and atf2 control the formation of a broad range of volatile esters. Appl. Environ. Microbiol. 2003, 69, 5228–5237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, M.J.; Bull, I.D.; Curnow, P. The yeast enzyme Eht1 is an octanoyl-CoA: Ethanol acyltransferase that also functions as a thioesterase. Yeast 2014, 31, 463–474. [Google Scholar] [PubMed] [Green Version]
- Dzialo, M.C.; Park, R.; Steensels, J.; Lievens, B.; Verstrepen, K.J. Physiology, ecology and industrial applications of aroma formation in yeast. FEMS Microbiol. Rev. 2017, 41, S95–S128. [Google Scholar] [CrossRef] [Green Version]
- Rauhut, D. Yeasts-production of sulfur compounds. Wine Microbiol. Biotechnol. 1993, 6, 183–223. [Google Scholar]
- Hirst, M.B.; Richter, C.L. Review of aroma formation through metabolic pathways of Saccharomyces cerevisiae in beverage fermentations. Am. J. Enol. Vitic. 2016, 67, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Dunn, B.; Levine, R.P.; Sherlock, G. Microarray karyotyping of commercial wine yeast strains reveals shared, as well as unique, genomic signatures. BMC Genom. 2005, 6, 53. [Google Scholar] [CrossRef] [Green Version]
- Henschke, P.; Jiranek, V. Yeasts-metabolism of nitrogen compounds. In Wine Microbiology and Biotechnology; Fleet, G., Ed.; Harwood Academic Publishers: Chur, Switzerland, 1993; pp. 77–164. [Google Scholar]
- Verstrepen, K.J.; Iserentant, D.; Malcorps, P.; Derdelinckx, G.; Van Dijck, P.; Winderickx, J.; Pretorius, I.S.; Thevelein, J.M.; Delvaux, F.R. Glucose and sucrose: Hazardous fast-food for industrial yeast? Trends Biotechnol. 2004, 22, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Gallone, B.; Steensels, J.; Prahl, T.; Soriaga, L.; Saels, V.; Herrera-Malaver, B.; Merlevede, A.; Roncoroni, M.; Voordeckers, K.; Miraglia, L.; et al. Domestication and divergence of Saccharomyces cerevisiae beer yeasts. Cell 2016, 166, 1397–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, K.S.; Klein, M.; Swiegers, J.H.; Hayasaka, Y.; Elsey, G.M.; Fleet, G.H.; Hoj, P.B.; Pretorius, I.S.; Lopes, M.A.D. Genetic determinants of volatile-thiol release by Saccharomyces cerevisiae during wine fermentation. Appl. Environ. Microbiol. 2005, 71, 5420–5426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossouw, D.; Naes, T.; Bauer, F.F. Linking gene regulation and the exo-metabolome: A comparative transcriptomics approach to identify genes that impact on the production of volatile aroma compounds in yeast. BMC Genom. 2008, 9, 530. [Google Scholar] [CrossRef] [Green Version]
- Richter, C.L.; Dunn, B.; Sherlock, G.; Pugh, T. Comparative metabolic footprinting of a large number of commercial wine yeast strains in chardonnay fermentations. FEMS Yeast Res. 2013, 13, 394–410. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-J.; Rathbone, D.; Asimont, S.; Adden, R.; Ebeler, S.E. Dynamic changes in ester formation during chardonnay juice fermentations with different yeast inoculation and initial brix conditions. Am. J. Enol. Vitic. 2004, 55, 346–354. [Google Scholar]
- Miller, A.C.; Wolff, S.R.; Bisson, L.F.; Ebeler, S.E. Yeast strain and nitrogen supplementation: Dynamics of volatile ester production in chardonnay juice fermentations. Am. J. Enol. Vitic. 2007, 58, 470–483. [Google Scholar]
- Steyer, D.; Ambroset, C.; Brion, C.; Claudel, P.; Delobel, P.; Sanchez, I.; Erny, C.; Blondin, B.; Karst, F.; Legras, J.L. QTL mapping of the production of wine aroma compounds by yeast. BMC Genom. 2012, 13, 573. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Rios, E.; Lopez-Malo, M.; Guillamon, J.M. Global phenotypic and genomic comparison of two Saccharomyces cerevisiae wine strains reveals a novel role of the sulfur assimilation pathway in adaptation at low temperature fermentations. BMC Genom. 2014, 15, 1059. [Google Scholar] [CrossRef] [Green Version]
- Saerens, S.M.G.; Delvaux, F.; Verstrepen, K.J.; Van Dijck, P.; Thevelein, J.M.; Delvaux, F.R. Parameters affecting ethyl ester production by Saccharomyces cerevisiae during fermentation. Appl. Environ. Microbiol. 2008, 74, 454–461. [Google Scholar] [CrossRef] [Green Version]
- Kruis, A.J.; Levisson, M.; Mars, A.E.; van der Ploeg, M.; Daza, F.G.; Ellena, V.; Kengen, S.W.M.; van der Oost, J.; Weusthuis, R.A. Ethyl acetate production by the elusive alcohol acetyltransferase from yeast. Metab. Eng. 2017, 41, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Kruis, A.J.; Gallone, B.; Jonker, T.; Mars, A.E.; van Rijswijck, I.M.H.; Wolkers-Rooijackers, J.C.M.; Smid, E.J.; Steensels, J.; Verstrepen, K.J.; Kengen, S.W.M.; et al. Contribution of eat1 and other alcohol acyltransferases to ester production in Saccharomyces cerevisiae. Front. Microbiol. 2018, 9, 3202. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.M.; Hong, E.L.; Amundsen, C.; Balakrishnan, R.; Binkley, G.; Chan, E.T.; Christie, K.R.; Costanzo, M.C.; Dwight, S.S.; Engel, S.R.; et al. Saccharomyces genome database: The genomics resource of budding yeast. Nucleic Acids Res. 2012, 40, D700–D705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallino, J.J.; Stephanopoulos, G. Metabolic flux distributions in Corynebacterium glutamicum during growth and lysine overproduction. Biotechnol. Bioeng. 1993, 41, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Varma, A.; Palsson, B.O. Metabolic capabilities of Escherichia coli: I. Synthesis of biosynthetic precursors and cofactors. J. Theor. Biol. 1993, 165, 477–502. [Google Scholar] [CrossRef]
- Pramanik, J.; Keasling, J.D. Stoichiometric model of Escherichia coli metabolism: Incorporation of growth-rate dependent biomass composition and mechanistic energy requirements. Biotechnol. Bioeng. 1997, 56, 398–421. [Google Scholar] [CrossRef]
- Pizarro, F.; Varela, C.; Martabit, C.; Bruno, C.; Prez-Correa, J.R.; Agosin, E. Coupling kinetic expressions and metabolic networks for predicting wine fermentations. Biotechnol. Bioeng. 2007, 98, 986–998. [Google Scholar] [CrossRef]
- Quiros, M.; Martinez-Moreno, R.; Albiol, J.; Morales, P.; Vazquez-Lima, F.; Barreiro-Vazquez, A.; Ferrer, P.; Gonzalez, R. Metabolic flux analysis during the exponential growth phase of Saccharomyces cerevisiae in wine fermentations. PLoS ONE 2013, 8, e71909. [Google Scholar] [CrossRef] [Green Version]
- Aceituno, F.F.; Orellana, M.; Torres, J.; Mendoza, S.; Slater, A.W.; Melo, F.; Agosin, E. Oxygen response of the wine yeast Saccharomyces cerevisiae EC1118 grown under carbon-sufficient, nitrogen-limited enological conditions. Appl. Environ. Microbiol. 2012, 78, 8340–8352. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Su, J.; Ma, W.; Guo, A.; Shan, Z.; Wang, H. Metabolic flux analysis of Saccharomyces cerevisiae in a sealed winemaking fermentation system. FEMS Yeast Res. 2015, 15. [Google Scholar] [CrossRef]
- Forster, J.; Famili, I.; Fu, P.; Palsson, B.O.; Nielsen, J. Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Res. 2003, 13, 244–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, N.C.; Herrgard, M.J.; Palsson, B.O. Reconstruction and validation of Saccharomyces cerevisiae iND750, a fully compartmentalized genome-scale metabolic model. Genome Res. 2004, 14, 1298–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nookaew, I.; Jewett, M.C.; Meechai, A.; Thammarongtham, C.; Laoteng, K.; Cheevadhanarak, S.; Nielsen, J.; Bhumiratana, S. The genome-scale metabolic model iIN800 of Saccharomyces cerevisiae and its validation: A scaffold to query lipid metabolism. BMC Syst. Biol. 2008, 2, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, M.L.; Palsson, B.Ø.; Herrgård, M.J. Connecting extracellular metabolomic measurements to intracellular flux states in yeast. BMC Syst. Biol. 2009, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Dobson, P.D.; Smallbone, K.; Jameson, D.; Simeonidis, E.; Lanthaler, K.; Pir, P.; Lu, C.; Swainston, N.; Dunn, W.B.; Fisher, P.; et al. Further developments towards a genome-scale metabolic model of yeast. BMC Syst. Biol. 2010, 4, 145. [Google Scholar] [CrossRef] [Green Version]
- Vargas, F.A.; Pizarro, F.; Pérez-Correa, J.R.; Agosin, E. Expanding a dynamic flux balance model of yeast fermentation to genome-scale. BMC Syst. Biol. 2011, 5, 75. [Google Scholar] [CrossRef] [Green Version]
- Nissen, T.L.; Schulze, U.; Nielsen, J.; Villadsen, J. Flux distributions in anaerobic, glucose-limited continuous cultures of Saccharomyces cerevisiae. Microbiology 1997, 143, 203–218. [Google Scholar] [CrossRef] [Green Version]
- Zomorrodi, A.R.; Maranas, C.D. Improving the iMM904 S. cerevisiae metabolic model using essentiality and synthetic lethality data. BMC Syst. Biol. 2010, 4, 178. [Google Scholar]
- Papp, B.; Szappanos, B.; Notebaart, R.A. Use of genome-scale metabolic models in evolutionary systems biology. Methods Mol. Biol. 2011, 759, 483–497. [Google Scholar]
- Lu, H.; Li, F.; Sanchez, B.J.; Zhu, Z.; Li, G.; Domenzain, I.; Marcisauskas, S.; Anton, P.M.; Lappa, D.; Lieven, C.; et al. A consensus S. cerevisiae metabolic model Yeast 8 and its ecosystem for comprehensively probing cellular metabolism. Nat. Commun. 2019, 10, 3586. [Google Scholar] [CrossRef] [Green Version]
- Heavner, B.D.; Price, N.D. Comparative analysis of yeast metabolic network models highlights progress, opportunities for metabolic reconstruction. PLoS Comput. Biol. 2015, 11, e1004530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heavner, B.D.; Smallbone, K.; Barker, B.; Mendes, P.; Walker, L.P. Yeast 5—An expanded reconstruction of the Saccharomyces cerevisiae metabolic network. BMC Syst. Biol. 2012, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, B.J.; Zhang, C.; Nilsson, A.; Lahtvee, P.-J.; Kerkhoven, E.J.; Nielsen, J. Improving the phenotype predictions of a yeast genome-scale metabolic model by incorporating enzymatic constraints. Mol. Syst. Biol. 2017, 13, 935. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Thiele, I.; Palsson, B.O. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Varma, A.; Palsson, B.O. Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli w3110. Appl. Environ. Microbiol. 1994, 60, 3724–3731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palsson, B. Systems Biology; Cambridge University Press: Cambridge, UK, 2015. [Google Scholar]
- Mahadevan, R.; Schilling, C.H. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 2003, 5, 264–276. [Google Scholar] [CrossRef]
- Becker, S.A.; Feist, A.M.; Mo, M.L.; Hannum, G.; Palsson, B.O.; Herrgard, M.J. Quantitative prediction of cellular metabolism with constraint-based models: The cobra toolbox. Nat. Protoc. 2007, 2, 727–738. [Google Scholar] [CrossRef]
- Schellenberger, J.; Que, R.; Fleming, R.M.T.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: The cobra toolbox v2.0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the cobra toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [Green Version]
- Droste, P.; Miebach, S.; Niedenführ, S.; Wiechert, W.; Nöh, K. Visualizing multi-omics data in metabolic networks with the software omix—A case study. Biosystems 2011, 105, 154–161. [Google Scholar] [CrossRef] [PubMed]
- King, Z.A.; Dräger, A.; Ebrahim, A.; Sonnenschein, N.; Lewis, N.E.; Palsson, B.O. Escher: A web application for building, sharing, and embedding data-rich visualizations of biological pathways. PLoS Comput. Biol. 2015, 11, e1004321. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, F. Über die bedingungen der fuselölbildung und über ihren zusammenhang mit dem eiweissaufbau der hefe. Ber. Dtsch. Chem. Ges. 1907, 40, 1027–1047. [Google Scholar] [CrossRef] [Green Version]
- Sentheshanmuganathan, S. The mechanism of the formation of higher alcohols from amino acids by Saccharomyces cerevisiae. Biochem. J. 1960, 74, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saerens, S.M.; Delvaux, F.R.; Verstrepen, K.J.; Thevelein, J.M. Production and biological function of volatile esters in Saccharomyces cerevisiae. Microb. Biotechnol. 2010, 3, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiropoulos, A.; Bisson, L.F. MET17 and hydrogen sulfide formation in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2000, 66, 4421–4426. [Google Scholar] [CrossRef] [Green Version]
- Dekoninck, T. Bottle Conditioning of Beer Strategies to Improve Yeast Refermentation Performance; Katholieke Universiteit Leuven: Leuven, Belgium, 2012. [Google Scholar]
- Aung, H.W.; Henry, S.A.; Walker, L.P. Revising the representation of fatty acid, glycerolipid, and glycerophospholipid metabolism in the consensus model of yeast metabolism. Ind. Biotechnol. 2013, 9, 215–228. [Google Scholar] [CrossRef] [Green Version]
- Dikicioglu, D.; Kirdar, B.; Oliver, S.G. Biomass composition: The “elephant in the room“ of metabolic modelling. Metabolomics 2015, 11, 1690–1701. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, A.; Beltran, G.; Warringer, J.; Guillamón, J.M. Genetic basis of variations in nitrogen source utilization in four wine commercial yeast strains. PLoS ONE 2013, 8, e67166. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, P.K.; Uppada, V.; Noronha, S.B. Comparison of pyruvate decarboxylases from Saccharomyces cerevisiae and komagataella pastoris (Pichia pastoris). Appl. Microbiol. Biotechnol. 2013, 97, 9439–9449. [Google Scholar] [CrossRef]
- Romano, P.; Suzzi, G.; Turbanti, L.; Polsinelli, M. Acetaldehyde production in Saccharomyces cerevisiae wine yeasts. FEMS Microbiol. Lett. 1994, 118, 213–218. [Google Scholar] [CrossRef]
- Rankine, B.C.; Bridson, D.A. Glycerol in australian wines and factors influencing its formation. Am. J. Enol. Vitic. 1971, 22, 6–12. [Google Scholar]
- Pigeau, G.M.; Inglis, D.L. Upregulation of ALD3 and GPD1 in Saccharomyces cerevisiae during icewine fermentation. J. Appl. Microbiol. 2005, 99, 112–125. [Google Scholar] [CrossRef]
- Malcorps, P.; Dufour, J.-P. Short-chain and medium-chain aliphatic-ester synthesis in Saccharomyces cerevisiae. Eur. J. Biochem. 1992, 210, 1015–1022. [Google Scholar] [CrossRef]
- Mason, A.B.; Dufour, J.-P. Alcohol acetyltransferases and the significance of ester synthesis in yeast. Yeast 2000, 16, 1287–1298. [Google Scholar] [CrossRef]
- Nordström, K. Studies on the Formation of Volatile Esters in Fermentation with Brewer’s Yeast. Ph.D. Thesis, Kungliga Tekniska Högskolan, Stockholm, Sweden, 1964. [Google Scholar]
- El-Sayed, A.M.; Heppelthwaite, V.J.; Manning, L.M.; Gibb, A.R.; Suckling, D.M. Volatile constituents of fermented sugar baits and their attraction to lepidopteran species. J. Agric. Food Chem. 2005, 53, 953–958. [Google Scholar] [CrossRef]
- Molina, A.M.; Swiegers, J.H.; Varela, C.; Pretorius, I.S.; Agosin, E. Influence of wine fermentation temperature on the synthesis of yeast-derived volatile aroma compounds. Appl. Microbiol. Biotechnol. 2007, 77, 675–687. [Google Scholar] [CrossRef]
- Birch, A.N.; Petersen, M.A.; Arneborg, N.; Hansen, Å.S. Influence of commercial baker’s yeasts on bread aroma profiles. Food Res. Int. 2013, 52, 160–166. [Google Scholar] [CrossRef]
- Thomas, D.; Surdin-Kerjan, Y. Metabolism of sulfur amino acids in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1997, 61, 503–532. [Google Scholar] [CrossRef]
- Shen, L.; Nishimura, Y.; Matsuda, F.; Ishii, J.; Kondo, A. Overexpressing enzymes of the ehrlich pathway and deleting genes of the competing pathway in Saccharomyces cerevisiae for increasing 2-phenylethanol production from glucose. J. Biosci. Bioeng. 2016, 122, 34–39. [Google Scholar] [CrossRef]
- Park, S.-H.; Kim, S.; Hahn, J.-S. Metabolic engineering of Saccharomyces cerevisiae for the production of isobutanol and 3-methyl-1-butanol. Appl. Microbiol. Biotechnol. 2014, 98, 9139–9147. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alcohol/Alcohol CoA | Ester | Organoleptic Properties | Present in Existing Model (iMM904) |
|---|---|---|---|
| Ethanol | Ethyl acetate | Fruity; solvent-like | Yes |
| 1-Propanol | Propyl acetate | Fruity; sweet | No, newly added |
| Isoamylol | Isoamyl acetate | Banana; pear | Yes |
| Isobutanol | Isobutyl acetate | Fruity; sweet | Yes |
| 2-Phenyl ethanol | 2-Phenylethyl acetate | Roses; honey; sweet | Yes |
| Tyrosol | Tyrosyl acetate | Floral; fruity | No, newly added |
| Methionol | Methionyl acetate | Sulfurous; cabbage; herbal | No, newly added |
| Hexa-1-ol | Hexyl acetate | Sweet; grape-like | No, newly added |
| Butanoyl-CoA | Ethyl butanoate | Banana; pineapple | No, newly added |
| Hexanoyl-CoA | Ethyl hexanoate | Aniseed; apple | No, newly added |
| Butanoyl-CoA | Ethyl butanoate | Banana; pineapple | No, newly added |
| Decanoyl-CoA | Ethyl decanoate | Waxy; fruity; sweet; apple | No, newly added |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scott, W.T., Jr.; Smid, E.J.; Notebaart, R.A.; Block, D.E. Curation and Analysis of a Saccharomyces cerevisiae Genome-Scale Metabolic Model for Predicting Production of Sensory Impact Molecules under Enological Conditions. Processes 2020, 8, 1195. https://doi.org/10.3390/pr8091195
Scott WT Jr., Smid EJ, Notebaart RA, Block DE. Curation and Analysis of a Saccharomyces cerevisiae Genome-Scale Metabolic Model for Predicting Production of Sensory Impact Molecules under Enological Conditions. Processes. 2020; 8(9):1195. https://doi.org/10.3390/pr8091195
Chicago/Turabian StyleScott, William T., Jr., Eddy J. Smid, Richard A. Notebaart, and David E. Block. 2020. "Curation and Analysis of a Saccharomyces cerevisiae Genome-Scale Metabolic Model for Predicting Production of Sensory Impact Molecules under Enological Conditions" Processes 8, no. 9: 1195. https://doi.org/10.3390/pr8091195