
Abstract
Background and Objectives
Lidocaine 5% medicated plaster is the first lidocaine containing product for chronic use. As no previous investigations have been conducted to evaluate the population pharmacokinetics of long-term exposure to lidocaine 5% medicated plasters, further insights into the evaluation of the pharmacokinetic properties of lidocaine and its metabolites were needed for the assessment of its safety.
Methods
The population pharmacokinetic properties of lidocaine and its metabolites were evaluated after multiple applications of lidocaine 5% medicated plasters based on data collected for up to 14.5 months from two phase III clinical trials (up to 2.5 months in the first trial, and up to 12 months in a follow-up trial) in post-herpetic neuralgia patients. Modeling was performed using nonlinear mixed effects as implemented in NONMEM® (nonlinear mixed-effect modeling) v.5. A stepwise forward inclusion and backward elimination procedure were used for covariate model building.
Results
The model provides reliable estimates of the pharmacokinetic behavior of lidocaine after medicated plaster application. It was validated using simulations and showed adequate predictive properties. Apparent Clearance was estimated to be 48 L/h after application of two or fewer plasters, whereas its value increased to 67 L/h after application of three plasters. Model-based simulations predicted no accumulation of lidocaine or any of its metabolites after long-term exposure of three simultaneous plasters up to 1 year. The variability explained by adding covariates into the model for the long-term exposures of lidocaine following one plaster or three simultaneous plaster applications was found to be very small with respect to the overall between-subject variability.
Conclusions
In conclusion, exposure to lidocaine after the application of the lidocaine medicated plaster was found to be primarily affected by the number of plasters simultaneously applied, i.e., it increased with the number of applied patches, but less than proportionally. No clinically relevant effect of other covariates was found to affect the exposure to lidocaine or its metabolites. As no accumulation was predicted by the model, long-term exposure to lidocaine and its metabolites is not expected to lead to any safety concerns in post-herpetic neuralgia patients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Population pharmacokinetic modeling shows that multiple applications of lidocaine 5% medicated plasters affect lidocaine pharmacokinetic properties. |
Exposure to lidocaine increases less than proportionally with an increasing number of plasters. |
1 Introduction
Post-herpetic neuralgia is a nerve pain, typically confined to one dermatome of the skin, following an infection with herpes zoster. It is the most common complication of herpes zoster [1] the incidence of which increases with age [2–4]. The pain is often described as burning, throbbing, aching, shooting, or stabbing, and may be constant or intermittent, spontaneous, or evoked [1, 5]. The patient’s quality of life is adversely affected [6, 7], with the condition often being underdiagnosed and undertreated [8].
Topical analgesic lidocaine 5% medicated plaster (Versatis®, Grünenthal GmbH, Aachen, Germany) is a hydrogel plaster containing 5% lidocaine. Each plaster contains 700 mg lidocaine, and up to three plasters can be applied simultaneously, but not overlapping, for up to 12 h every 24 h. The plaster has been approved for the symptomatic relief of neuropathic pain associated with a previous herpes zoster infection in 50 countries and in 12 of these countries for the treatment of localized peripheral neuropathic pain [9–11]. The broad level of clinical evidence for its use in localized neuropathic pain has recently been reviewed [12]. It has also been recommended as a first line treatment, especially in frail and elderly patients when there are concerns regarding side effects or safety of other treatments [11].
The lidocaine 5% medicated plaster is easy to use and does not require titration, in contrast to systemic medications. Its use is associated with good short- and long-term tolerability and a low incidence of systemic adverse drug reactions [13].
Lidocaine has a long history of use in humans. Initial doses of 1.5 mg/kg within 2–4 min are given intravenously up to three times within 1 h for the initial treatment of ventricular arrhythmias. Infusions of 4 mg/min/70 kg are administered for up to 24 h to maintain normal sinus rhythm. Plasma concentrations of 1500–5000 μg/L are reported for this dosing regimen. In clinical trials, continuations of lidocaine infusions for several days have been reported [14–16].
However, this therapy is not suitable for ongoing chronic pain. Lidocaine 5% medicated plaster was, therefore, developed to provide a formulation that makes lidocaine topically available to the dermal layers of the skin. Further insights into the evaluation of the pharmacokinetic properties of lidocaine and its metabolites were needed for assessment of its safety, e.g., for use in patients with mild-to-moderate cardiac or hepatic dysfunction.
Lidocaine is metabolized rapidly by the liver to a number of metabolites, including monoethylglycinexylidide (MEGX) and glycinexylidide (GX), both of which have pharmacologic activity although with less potency than that of lidocaine [17]. These metabolites are further metabolized to 2,6-xylidine. The amount of lidocaine systemically absorbed from the plaster is related to both the duration of the application and the surface of the body to which it is applied. The penetration of lidocaine into intact skin after application of lidocaine 5% medicated plaster is sufficient to produce a local analgesic effect, but less than necessary to produce loss of sensation and numbness.
The pharmacokinetics of lidocaine 5% medicated plaster has been evaluated in healthy volunteers after the simultaneous application of four plasters for three consecutive days changed every 12 or 24 h [18], after the simultaneous application of four plasters for 18 h/day for three consecutive days [19] and after the simultaneous application of three plasters for 12 h/day, repeated for three periods which were separated by 12 h “rest period” [20]. In this last study, systemic exposure of lidocaine and MEGX was also assessed in acute herpes zoster patients and post-herpetic neuralgia patients after the single application of three plasters. In healthy subjects, mean C max after single application was 128 ± 63 μg/L (mean ± SD [standard deviation]). Maximum lidocaine plasma concentrations in patients with herpes zoster and patients with post-herpetic neuralgia were substantially lower with 74 ± 38 and 52 ± 31 μg/L (mean ± SD), respectively. Based on this study, when lidocaine 5% medicated plaster is used according to the maximum recommended dose (three plasters applied simultaneously for 12 h), about 3 ± 2% of the total applied lidocaine dose is systemically available and similar for single and multiple administrations.
Population pharmacokinetics utilizes sparsely collected drug concentration data to identify covariate effects of pathophysiologic factors (e.g., body weight, excretory, and metabolic functions) possibly requiring changes in the dosing regimen [21].
Up to now, no population pharmacokinetic assessment of the exposure to lidocaine and its metabolites after long-term therapy with lidocaine 5% medicated plaster has been performed. Given that lidocaine 5% medicated plaster is the first lidocaine containing product for chronic use, further insights into the pharmacokinetic properties of lidocaine and its metabolites were needed for the assessment of its safety.
The objectives of this study were to evaluate the population pharmacokinetic properties of lidocaine and MEGX, GX, and 2,6-xylidine after application of lidocaine 5% medicated plaster based on data from two phase III clinical trials in patients suffering from post-herpetic neuralgia, to estimate the population exposure to lidocaine after multiple plaster applications, and to explore the influence of covariates on the exposure to lidocaine and its metabolites.
2 Materials and Methods
2.1 Materials
Medicated plasters were manufactured by Teikoku Seiyaku Company, Japan.
Each plaster contained 700 mg lidocaine as the drug substance, and methyl-parahydroxybenzoate and propyl-parahydroxybenzoate as preservatives in an aqueous base.
The lidocaine 5% medicated plaster comprised an adhesive material containing lidocaine applied at 5 mg/cm2 to a non-woven polyester felt backing and covered with embossed film. The plasters measured 14 cm × 10 cm. Each gram of adhesive in lidocaine 5% medicated plaster contained lidocaine 50 mg, in a base of purified water, glycerol, d-sorbitol, sodium polyacrylate, sodium carboxymethylcellulose, propylene glycol, urea, polyacrylic acid, kaolin, tartaric acid, gelatin, polyvinyl alcohol, dihydroxyaluminum aminoacetate, disodium edetate, methyl-parahydroxybenzoate, and propyl-parahydroxybenzoate.
2.2 Trials
Two trials contributed patients to this investigation:
The first trial [22] was a double-blind, multicenter, multiple-dose, enriched-enrolment, randomized-withdrawal, parallel-group phase III trial with lidocaine 5% medicated plaster and corresponding placebo plaster in patients suffering from post-herpetic neuralgia. The trial was designed as an enriched-enrollment, randomized-withdrawal trial consisting of an open-label run-in phase of 8 weeks followed by a withdrawal phase of up to 2 weeks. The trial included male and female patients aged 50 years and older, suffering from post-herpetic neuralgia and having an average pain intensity (during last week prior to screening and enrolment visit) of at least 4 on the 11-point numeric rating scale (NRS; scale of 0–10). Post-herpetic neuralgia was defined as neuropathic pain persisting for at least 3 months after healing of a herpes zoster skin rash.
The second trial [23] was an open-label, multicenter, multiple-dose, phase III long-term trial of 12 months with lidocaine 5% medicated plaster in patients aged 50 years and older suffering from post-herpetic neuralgia. Patients were offered the option of entering this second trial if they had completed the first trial or when enrolment in the double-blind withdrawal phase of the first trial was stopped, because the planned number of 70 randomized patients was attained. Once the randomized-withdrawal trial had been completed, other patients were allowed to enter the second trial directly to reach a planned number of approximately 100 patients with treatment over a period of at least 6 months to fulfill regulatory requirements.
Both trial protocols were approved by regional ethics committees and competent national authorities.
2.3 Patients
The patient population included in this pharmacokinetic analysis consisted of patients for whom at least one concentration of either lidocaine, MEGX, GX, or 2,6-xylidine was available with the appropriate dosing and sampling history. The pooled pharmacokinetic database included 212 patients with contributing pharmacokinetic information. The demographic characteristics of the patients in the pooled pharmacokinetic database are given in Table 1.
2.4 Drug Administration
In the first trial, patients applied the lidocaine 5% medicated plasters for up to 10 weeks. The plasters were applied topically. Patients could apply up to three plasters at a time. They had to wait for at least 12 h before it was allowed to apply further plasters. The maximum application duration for each plaster was 12 h.
In the second trial, patients applied the lidocaine 5% medicated plaster for a maximum of 12 months. The patient could apply the plasters during the day or night, but only for a maximum of 12 h during a 24-h period.
2.5 Blood Sampling
A sparse sampling scheme was used to lessen the burden on the patients. Blood samples were drawn for the evaluation of systemic exposure to lidocaine and its metabolites on up to five occasions per patient in each of the trials.
In the first trial, blood samples were taken during enrolment without medication, at week 1, week 4, and week 10 with the plaster in situ, and at week 8 with and without the plaster in situ.
In the second trial, blood samples were taken at week 12, week 26, and week 42 with the plaster in situ, and at week 52 with and without the plaster in situ. An additional sample was taken at enrolment for patients not recruited from the first trial.
Blood samples were handled as follows: approximately 40–60 min after collection, the blood samples were centrifuged at 3500–4000 rpm at 18 °C for 5 min. The serum was transferred into a fresh uncoated serum tube with a volume of about 4 to 5 mL and stored at or below −17 °C until shipment to a central laboratory on dry ice.
Concentration data were available from 212 patients comprising 1989 concentrations (Table 2, number of plasters versus observed lidocaine concentrations).
2.6 Bioanalysis
The concentrations of lidocaine and its metabolites MEGX, GX, and 2,6-xylidine were determined in serum samples after ultrafiltration by a validated liquid chromatography-tandem mass spectrometry (LC–MS/MS) method at A&M, Labor für Analytik und Metabolismusforschung Service GmbH, Kopernikusstrasse 25, 50126 Bergheim, Germany. Quantification was performed using an internal standard method. Calibration of lidocaine in human serum was performed in the range of 0.500–300 ng/mL. Calibrations of MEGX and GX were performed in the range of 0.500–300 or 150 ng/mL and of xylidine in the range of 0.500–300 and 108 ng/mL. The lower limit of quantification (LLOQ) for all four analytes was approximately 0.500 ng/mL.
The calculated inter-assay accuracy and inter-assay precision of the calibration and quality control samples were within the accepted ranges for bioanalytical methods.
2.7 Population Pharmacokinetic Modeling Analyses
Pharmacokinetic data were evaluated by means of a population pharmacokinetic modeling approach as implemented in the NONMEM program (v.5 level 1.1) with visual NM as the Windows interface and the Digital Visual Fortran v5.0 compiler. Data handling for representation and summary statistics was performed using SAS v8.2, and graphs were made with SigmaPlot v8.
The modeling strategy of this study included two sequential steps: first, a base structural model for the concentrations of lidocaine and its metabolites was developed; this model was subsequently followed by a covariate model development and final model selection by covariate forward inclusion and backward deletion from the full model.
2.7.1 Model Assumptions
All parameters were assumed to be log-normally distributed as given in
where
-
\(\theta_{i}\) is the estimated parameter value for individual i.
-
\(\theta_{\text{TV}}\) is the typical population value (geometric mean) of the parameter.
-
\(\eta_{i}\) is the inter-individual variability (IIV) which is assumed to follow a normal distribution with mean of zero and variance of \(\omega^{2}\).
The magnitude of IIV was expressed as coefficient of variation (%CV), which was approximated by the square root of the variance estimate.
The residual error model was described by an additive error model as in
where
-
C ij is the jth measured observation in individual i.
-
\(\hat{C}_{ij}\) is the jth model-predicted value in individual i.
-
\(\varepsilon_{aij}\) is the additive residual random error for individual i and measurement j and it is assumed to be normally distributed with mean of zero and variance of \(\sigma^{2}\).
Alternative error models, such as proportional and combined error models, were also tested during model development.
2.7.2 Criteria for Model Selection
Pre-defined criteria were used to select a model among possible models. Successful minimization runs were needed, and goodness-of-fit plots were produced, including population and individual predictions versus observations, weighted residuals versus population predictions and versus time post-dose.
The addition of a pharmacokinetic parameter was based on the decrease of the objective function: for one additional parameter, the decrease was to be at least 3.84, which approximates a p < 0.05, assuming a χ 2 distribution of the objective function value (OFV), i.e., −2log (likelihood).
2.7.3 Structural Pharmacokinetic Model
Model construction started by fitting lidocaine and 2,6-xylidine data first. Once an acceptable structural model had been established for these analytes, two additional compartments were added to accommodate the two additional metabolite concentrations. The selection of the rate constants connecting compartments was initially guided by the known metabolic pathways. The best model for the four analytes with statistical components describing inter-individual variability and residual error constituted the base structural model.
2.7.4 Inclusion of Covariates
Extensive exploratory analysis of the variables selected as potential covariates was performed using summary statistics, plots of correlation, and histograms of their distribution. The covariate selection was performed with NONMEM using the Likelihood Ratio Test. Once the base model had been identified, each covariate was introduced separately in the model and its effect on the OFV determined its significance: a decrease of the OFV of at least 3.84 was required for one additional parameter to be declared significant (p < 0.05). The selected covariate effects were ranked according to the decrease in OFV they caused. The model was then expanded by including identified significant covariate effects, one after the other, starting with the strongest (largest change in OFV [ΔOFV]). Each model extension was tested according to the acceptance criteria and the Likelihood Ratio Test (p < 0.05). These steps lead to the most complex model, the full model, which was subsequently tested for redundancies by removing covariate effects one at the time starting with the weakest, i.e., using backward deletion. Effects were declared redundant and removed from the model according to the Likelihood Ratio Test at p < 0.01 or ΔOFV < 6.63.
The effect of the following covariates was tested:
-
Total bilirubin (BIL) and cardiac insufficiency on volumes of distribution.
-
Age, weight, height, sex, trial, systolic and diastolic blood pressure, body mass index (BMI), lean body mass, body surface area, body fat, and smoking status on all model parameters.
-
Creatinine clearance (CLCR) and heart rate on absorption and elimination.
-
Aspartate transaminase (AST), alanine transaminase (ALT), creatine kinase (CK), lactate dehydrogenase (LDH), total BIL, gamma-glutamyltransferase (gamma-GT), concomitant medication, serum creatinine, and urine pH on elimination.
The effect of concomitant medications, such as atenolol (ATEN), a cytochrome P450 1A2 (CYP1A2) substrate, and metoprolol and beta-blockers, as separate group, was explored to test their potential influence on any of the pharmacokinetic parameters. Concomitant medication was expressed as binary data (concomitant medication yes or no). The laboratory safety parameters were used as dichotomous covariates.
The influence of binary covariates, e.g., concomitant medications, on the typical population value of a parameter (\(\theta_{\text{TV}}\)), was modelled as follows:
where θ 1 represents the parameter value in subjects receiving concomitant medication and θ 2 is the parameter value in subjects not receiving the concomitant medication. The use of a concomitant medication was coded as 1 for the subjects who received it and as 0 for the subjects who did not receive it.
2.7.5 Model Evaluation
Any deviation from the model selection and acceptance criteria was discussed. Goodness-of-fit plots were produced, including population and individual predictions versus observations, weighted residuals versus population predictions and versus time post-dose, on linear and logarithmic scales. For the final model, the distribution of the random effects around 0 and potential correlations between them were checked.
The fixed-effect parameters were reported associated with a 95% confidence interval obtained from the NONMEM reported standard error of the estimates (SEE) and the population value of that parameter, θ ± 1.96 × SEE. To evaluate the performances of the model in predicting lidocaine and metabolites concentrations, the final model was subjected to the predictive performance check (PPC).
Three subsets of concentration–time data were produced for each analyte:
-
1.
Time class 1: time after start of treatment <2000 h.
-
2.
Time class 2: time after start of treatment from 2000 to <8000 h.
-
3.
Time class 3: time after start of treatment >8000 h.
For estimating the predictive performance of the model, 200 replicates of the data subsets were simulated using NONMEM. The simulated concentrations stored in the dependent variable array were uploaded into an SAS data set to derive, for each observed concentration, i.e., each analyte at any one time point, the 5th and 95th percentile of the simulated data.
For each subset, the number and percentage of observed concentrations within this predicted 90% range were calculated. The model passed the check when about 90% of the observations fell within the range.
In addition, the predictive power of the model was assessed by mean of a visual predictive check (VPC). The VPC was based on the simulations obtained from the final model (n = 500). The results of the visual predictive check were summarized graphically.
2.7.6 Simulations
To interpret the effect of dose (dose proportionality), pharmacokinetic (PK) covariates (subgroups of subjects), and time in the trial (accumulation), the final model was used to perform population predictions and Monte Carlo simulations of the lidocaine population exposures. Under the assumption that the plasters released drug at a constant rate as described for the population pharmacokinetic analysis, simulations were performed for the first day, the fourth day, and for 1 year of treatment upon application of one plaster or upon the simultaneous application of three plasters. The plaster or plasters were applied for only 12 h in each 24 h dosing interval.
The parameter estimates of the final model were used for the simulations. NONMEM was used to simulate the virtual data ($SIM ONLY). R 3.0.1 was used to read the simulated data sets to derive percentiles of the distributions and to export percentiles as comma separated values (csv) files. Figures presenting the simulated systemic exposure were generated with R 3.0.1.
2.7.7 Assumptions
The following assumptions were applied throughout the analysis:
-
1.
Lidocaine 5% medicated plasters were assumed to deliver lidocaine at a constant rate (i.e., 1800 µg/h).
-
2.
Missing covariate data per patient was replaced by the mean of the missing covariate in the specific subgroup.
-
3.
Values for covariates were constant over the entire observation time.
-
4.
If the number of concentrations below the limit of quantification (LOQ) was <10%, then measurements of concentrations below LOQ were set to ½ × LOQ.
-
5.
All co-medications, independent of start or stop dates, were assumed to be in use throughout the observation period of the trial.
3 Results
Modeling started with the development of a structural model. The primary aim of the structural model was to adequately describe the likely concentrations to be observed after application of up to three simultaneous plasters onto the skin in the target population while incorporating some aspects of the known or hypothesized pathways of lidocaine metabolism [24].
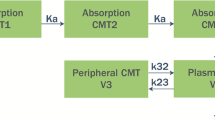
This model is a linear model of four compartments, one for each chemical entity. The ADVAN 5 subroutine in NONMEM, which implements a user-defined general linear model, was used.
Since lidocaine metabolites were not administered alone and the true fraction of lidocaine converted to its metabolites is unknown, the fraction of lidocaine converted to its metabolites and the apparent volume of distribution of the metabolites are unidentifiable in the model. Hence, the apparent volumes of distribution of the metabolites were fixed to an arbitrary value (100 L) [25, 26].
Inter-individual variabilities were tested and added on the apparent volume of distribution of compartment 1 (lidocaine), V 1, the elimination rate constant of compartment 3 (GX), k 30, and the elimination rate constant of compartment 4 (2,6-xylidine), k 40. The inter-individual variability was best described by the exponential model, and the residual error structure was best described by an additive error model. Residual error represents the differences between the observed and individual-predicted concentrations, and included within-patient variability, assay errors, and model misspecification errors. The base model was evaluated by means of goodness-of-fit plots and PPCs. The PPCs demonstrated that the base model-predicted concentrations of lidocaine and its metabolites equally well. It was, therefore, decided to proceed with covariate selection and to develop a full model.
The first expansion of the base model investigated the influence of trial design parameters (DLVL = Dose level, i.e., number of plasters applied simultaneously, TIME after start of treatment, and FORM, a binary covariate identifying the two trials combined in the data set) on the pharmacokinetic parameters affected by inter-individual variabilities. Among them, the effect of DLVL >2 on k 30 and on V 1 was the strongest. The influence of DLVL can be interpreted as a decrease of systemic bioavailability when the dose, i.e., the number of plasters, increases.
Several covariates were tested on the same pharmacokinetic parameters for which inter-individual variabilities were estimated and they included body size, age, enzyme activities, renal, hepatic, cardiovascular status, and information on co-medication.
During the forward inclusion, the following covariates were found significant: DLVL on V 1, ATEN, BIL, DLVL, ALT, CYP1A2 substrate, CLCR, BMI on k 30, and LDH and ALT on k 40. During the backward deletion, only ATEN was found of no significance in the final model. The DLVL on k 30 was still found to have the strongest influence on the OFV among all covariates.
CLCR was found to be at the limit of statistical significance (ΔOFV = 6.72). Since CLCR, however, describes the relationship between excretion of GX and kidney function, which naturally decreases with increasing age, the parameter was kept in the final model.
The model did not detect any change in pharmacokinetic parameters when the duration of the treatment increased.
The estimates of between-subject variability (CVs) associated with k 30, k 40, and V 1 were 62.4, 44.7, and 55.9%, respectively. After inclusion of PK covariates, the between-subject variability has decreased by 17.5% for k 30, 8.3% for k 40, and increased by 4.4% for V 1 from the base model. The residual error on each of the four analytes has decreased only moderately between the base and the final model, mainly for GX concentrations.
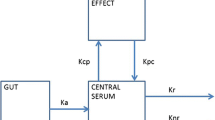
The resulting final model is depicted in Fig. 1, and Table 3. Parameter estimates of the final model lists the parameter estimates.

Graphical representation of the final model. ALT alanine transaminase, BIL bilirubin, BMI body mass index, CL CR creatinine clearance, DLVL dose level, i.e., number of plasters applied simultaneously, GX glycinexylidide, k 30 first-order rate constant from compartment 3 to the outside, k 40 first-order rate constant from compartment 4 to the outside, k 12 first-order rate constant from compartment 1 to compartment 2, k 14 first-order rate constant from compartment 1 to compartment 4, k 23 first-order rate constant from compartment 2 to compartment 3, LDH lactate dehydrogenase, MEGX monoethylglycinexylidide, CYP1A2 substrate substrate of cytochrome 1A2 isozyme, V 1 volume of distribution of compartment 1, V 2 volume of distribution of compartment 2, V 3 volume of distribution of compartment 3, V 4 volume of distribution of compartment 4
As the table shows, the factor which mostly affects lidocaine pharmacokinetics is the number of plasters applied simultaneously. The model predicts less exposure than predicted from linear kinetics, affecting the volume of distribution of lidocaine and the rate constant of elimination of the metabolite GX.
The apparent volume of distribution of lidocaine is about 19 and 26 L/kg (assuming a typical subject of 70 kg) after the application of two or fewer plasters and after the application of three plasters, respectively.
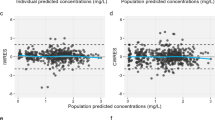
The final model was evaluated by means of goodness-of-fit plots. As shown in Fig. 2, no particular bias can be detected in the model. The outliers (points outside the ±2 interval) visible in Fig. 2c were kept in the final model.

Lidocaine goodness-of-fit plot for final model (a) population predicted concentrations of lidocaine vs. observations; (b) individual-predicted concentrations of lidocaine vs. observations; and (c) conditional weighted residuals vs. time. The red line represents the identity line, and the blue line represents the smooth line. CWRES conditional weighted residuals, DV dependent variable, PRED population predicted concentrations, IPRED individual-predicted concentrations
The results of the PPC of the final model indicate that there is no difference in the model performance for different time periods after initiation of therapy and the model predicts all four types of concentrations equally well, as displayed in Table 4, predictive performance check (PPC) of the final model. About 90% of observations lie within the 5th to 95th percentile range of the simulated predictions.
The predictive power of the model was assessed by means of VPC (Fig. 3). As shown in the figure, the model is able to capture the general trend of the data adequately, although it slightly overestimates the median. The upper bound of the 90% prediction interval appears to be very well defined.

Visual predictive check of the final model (n = 500 simulations)
To elucidate the effect of the dose on the exposure, i.e., the effect of a decrease in systemic bioavailability when the dose (the number of plasters) increases, simulations of lidocaine population concentrations were performed for the first day and the fourth day of treatment, after application of 1 or three plasters, in 500 individuals. Plaster administration duration was assumed to be 12 h. The results are displayed in Fig. 4. As shown in the figure, according to the final pharmacokinetics model, maximum serum concentrations (C max) increase less than proportionally to the dose level. Table 5 summary of the simulated values of C max after the first and fourth doses of treatment with one plaster, or three medicated plasters displays the summary statistics of the simulated C max values.

Comparison of simulated C max values after the first and fourth doses of treatment with (a) one plaster or (b) three medicated plasters. C max maximum concentration
Simulations were also performed to quantify the effects of pharmacokinetic covariates. For this purpose, Monte Carlo simulations of the time course of concentrations over a 24-h dosing interval with the application of one and three lidocaine plasters were made for a population of 200 subjects not affected by the pharmacokinetic covariates co-administration of CYP1A2 substrates and by laboratory safety parameters from the 30th percentile, compared to 200 subjects with pharmacokinetic parameters affected by pharmacokinetic covariates.
Figure 5 shows percentiles of the population without affecting covariates (in different shades of grey), superimposed with continuous lines representing the same percentiles for the population with covariates affecting the pharmacokinetics. In the figure, it can be seen that the influence of subject factors is very modest that the change in exposure is very small and much smaller than between-subject variability.

Simulated exposure to lidocaine of typical subjects: (a) exposure after 1 year of treatment with one plaster applied for 12 h every day and (b) exposure after 1 year of treatment with three plasters applied for 12 h every day. Solid black lines indicate 5th, 25th, 50th, 75th, and 95th percentiles of subjects with covariates
The effect of time in the trial was also assessed by means of simulations. The simulated population exposure of lidocaine and its metabolites after the application of three plasters simultaneously to subjects without the influence of covariates for the first 8 days of treatment and after one year (Fig. 6) indicates that steady-state conditions are reached for lidocaine and its metabolites on the fourth day of treatment and that no accumulation is predicted afterwards.

Simulated population exposure to lidocaine and its metabolites after simultaneous application of three plasters up to 1 year. GX glycinexylidide, MEGX monoethylglycinexylidide
4 Discussion
In the current analysis, a parent-metabolite model able to describe the pharmacokinetics of lidocaine and its metabolites after long-term application of up to three lidocaine 5% medicated plasters for a maximum period of 12 h in post-herpetic neuralgia patients has been developed. Up to now, this is the first model-based evaluation of long-term exposure to lidocaine 5% medicated plaster.
The results of this model-based analysis confirm previous findings on the low bioavailability of lidocaine after topical application. Given the model-based estimate of apparent volume of distribution after the application of three plasters (26 L/kg) and the reported volume of distribution after intravenous administration (1.3 L/kg) [27], it was possible to compute the apparent systemic bioavailability after the application of three plasters based on the following equation:
where V TOP is the apparent volume of distribution after the topical application of the plaster, V IV is the volume of distribution after intravenous administration, and F is the apparent systemic bioavailability of lidocaine after topical application of the plaster.
Based on our model, the apparent systemic bioavailability for lidocaine after the application of three plasters is 1.3–26 L/kg = 0.05, i.e., 5%, which is consistent with the reported value of 3 ± 2% from a previous study [20], and which confirms the low extent of absorption of lidocaine into the systemic circulation from the plasters. We are aware that the values of volume of distribution used in this calculation derive from different studies and different patient groups; therefore, this should be considered as an approximation of the level of systemic bioavailability after the application of lidocaine 5% medicated plaster.
One of the main findings of our investigation is the fact that the apparent clearance of lidocaine is dependent on the number of plasters applied simultaneously. Based on the final parameter estimates, the apparent total clearance of the drug from plasma after dermal administration (CL/F) could be calculated based on the following equation:
where K el is the elimination rate constant of lidocaine after dermal administration.
As the apparent volume of distribution was found to be 1320 L after the application of two or fewer plasters and 1810 L after the application of three plasters and lidocaine K el was estimated at 0.0037 h−1 (sum of k 12 and k 14), the apparent clearance was computed as 48.8 L/h after the application of two or fewer plasters and 67.0 L/h after the application of three plasters.
The effect of dose on the exposure has been further investigated by means of simulations. As shown in Fig. 4 and Table 5. Summary of the simulated values of C max after the first and fourth doses of treatment with one plaster, or three medicated plasters, the maximum concentrations of lidocaine clearly increase less than proportionally to the dose level and remain well below the level associated with toxicity (>6000 μg/L). These findings confirm the safe profile of lidocaine 5% medicated plaster when more than two plasters are applied simultaneously.
Several covariates were tested in this population pharmacokinetics analysis. DLVL, BIL, ALT, CYP1A2 substrate, CLCR, and BMI were found to be statistically significant. In a population pharmacokinetic analysis, it is crucial to assess if the covariates found to be statistically significant in the final model are also clinically relevant, i.e., if a dose adjustment based on these covariates needs to be considered. In the current model, the effect of BMI on the elimination half-life of GX, k 30, indicated a modest decrease for patients with a BMI >27.9 kg/m2, which could not be explained by any physiological mechanism. Analogously, when patients are treated with concomitant substrates of the CYP1A2, the model predicts a decrease of the elimination half-life.
Theoretically, given the involvement of CYP1A2 in lidocaine’s metabolism, this effect could be explained by the assumption that the concomitant substrate is a strong enzyme inhibitor.
However, the confidence interval for the effect of CYP1A2, as well as for the effects of LDH and ALT, was found to be quite large. Therefore, these effects might be due to spurious effects emerging from a large number of tests, rather than to a real biological explanation. To assess whether these covariates might indeed be clinically relevant, Monte Carlo simulations of the time course of concentrations over a 24-h dosing interval with the application of lidocaine plasters were performed for a population of subjects not affected by the pharmacokinetic covariates, compared to subjects with pharmacokinetic parameters affected by pharmacokinetic covariates (Fig. 5).
Clearly, the influence of subject factors is very limited and the change in exposure is very small and much smaller than between-subject variability. Based on this finding, no dose adjustment of dermal lidocaine might need to be considered for specific patient subgroups.
A previous investigation on lidocaine 5% medicated plaster [20] indicated that no accumulation of lidocaine was observed after repeated doses for up to 3 days, while some accumulation of the MEGX metabolite was observed from day 1 to day 2. In our study on lidocaine 5% medicated plaster, for the first time, long-term exposure to lidocaine plaster has been evaluated. The present data extend up to 18 months of treatment for some subjects and up to 1 year for most subjects. Based on our model-based simulations, steady-state conditions are reached at the 4th day of treatment and no accumulation is predicted after steady state is reached for lidocaine or for any of its metabolites (Fig. 6). Furthermore, the duration in the trial was shown not to affect any pharmacokinetic parameter with inter-individual variability, i.e., V 1, k 30, and k 40. The investigation from Gammaitoni et al. [19] revealed that steady-state plasma conditions were achieved within 3 days of application of four plasters for 18 h/day in healthy volunteers. In our study, steady-state conditions might have been achieved slightly later given the different study population.
4.1 Limitations
The sparse nature of the data available in this investigation did not support the simultaneous estimation of the covariate effects and of the rate constants of biotransformation of lidocaine into MEGX (k 12) and into 2,6-xylidine (k 14) in the full model. To avoid high correlations among parameter estimates, k 12 and k 14 were fixed to values similar to those estimated during covariate forward inclusion (0.003 and 0.007 h−1, respectively). Since in a sensitivity analysis which was conducted to estimate the effect of changing the values of these parameters all the runs converged without a change in objective function, the modeling option of using fixed values for k 12 and k 14 was accepted.
The VPC in Fig. 3 shows that the model slightly overestimates the median of lidocaine concentrations. As the focus of our analysis is on the potential safety concerns due to lidocaine exposure, we believe that the model overestimation does not impact the conclusion of our analysis on lidocaine safety profile.
We are aware about the presence of additional variability which cannot be explained by the inter-individual variability in our model. As shown in Fig. 2b (correlation between individual predictions and observations), some values appear not to be uniformly distributed along the line of identity, which could also be interpreted as an indication that the model was not “over fitted” and that shrinkage is between acceptable boundaries. The unexplained variability can be due to several factors, i.e., topical administration and compliance issues linked to unsupervised plaster applications. Despite such limitations and taking into account the complexity of long-term applications, the sparse nature of the data and that only the parent drug but not the metabolites could be dosed, we believe that the current model is able to describe reasonably well both parent and metabolite data.
5 Conclusions
The data collected in 212 subjects with post-herpetic neuralgia treated with lidocaine 5% medicated plaster for up to 14.5 months were used to build a pharmacokinetic model to predict concentrations of lidocaine and three metabolites simultaneously.
The analysis resulted in the estimation of a model that provided reliable estimates of the pharmacokinetic behavior of lidocaine after the lidocaine 5% medicated plaster application. The model was validated using simulations and showed reliable predictive properties.
The model demonstrated that exposure to lidocaine is primarily affected by the number of plasters simultaneously applied and that it increases less than proportionally with an increasing number of plasters. The modest effects of safety parameters, such as CLCR, BIL, ALT, and LDH as well as BMI and concomitant medication on the pharmacokinetic model’s parameters, although statistically significant, were of no clinical relevance. The effect of covariates on the long-term exposures of lidocaine after one plaster or three simultaneously applied plasters was found very small with respect to between-subject variability of exposure.
As no accumulation was predicted by the model, long-term exposure to lidocaine and its metabolites is not expected to lead to any safety concern in post-herpetic neuralgia patients.
References
Johnson RW, McElhaney J. Postherpetic neuralgia in the elderly. Int J Clin Pract. 2009;63(9):1386–91.
Yawn BP, Saddier P, Wollan PC, et al. A population-based study of the incidence and complication rates of herpes zoster before zoster vaccine introduction. Mayo Clin Proc. 2007;82(11):1341–9.
Gauthier A, Breuer J, Carrington D, et al. Epidemiology and cost of herpes zoster and post-herpetic neuralgia in the United Kingdom. Epidemiol Infect. 2009;137(1):38–47.
Gialloreti LE, Merito M, Pezzotti P, et al. Epidemiology and economic burden of herpes zoster and post-herpetic neuralgia in Italy: a retrospective, population-based study. BMC Infect Dis. 2010;10:230.
Dworkin RH, Gnann Jr JW, Oaklander AL, et al. Diagnosis and assessment of pain associated with herpes zoster and postherpetic neuralgia. J Pain. 2008;9(1 Suppl 1):S37–44.
Johnson RW, Bouhassira D, Kassianos G, et al. The impact of herpes zoster and post-herpetic neuralgia on quality-of-life. BMC Med. 2010;8:37.
Lukas K, Edte A, Bertrand I. The impact of herpes zoster and post-herpetic neuralgia on quality of life: patient-reported outcomes in six European countries. Z Gesundh Wiss. 2012;20(4):441–51.
Nalamachu S, Morley-Forster P. Diagnosing and managing postherpetic neuralgia. Drugs Aging. 2012;29(11):863–9.
Dworkin RH, O’Connor AB, Audette J, et al. Recommendations for the pharmacological management of neuropathic pain: an overview and literature update. Mayo Clin Proc. 2010;85(3 Suppl):S3–14.
Attal N, Cruccu G, Baron R, et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol. 2010;17(9):1113–88.
Finnerup NB, Attal N, Haroutounian S, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 2015;14(2):162–73.
de León-Casasola OA, Mayoral V. The topical 5% lidocaine medicated plaster in localized neuropathic pain: a reappraisal of the clinical evidence. J Pain Res. 2016;9:67–79.
Navez ML, Monella C, Bösl I, et al. 5% lidocaine medicated plaster for the treatment of postherpetic neuralgia: a review of the clinical safety and tolerability. Pain Ther. 2015;4(1):1–15.
Benowitz NL, Meister W. Clinical pharmacokinetics of lignocaine. Clin Pharmacokinet. 1978;3(3):177–201.
Estes NA 3rd, Manolis AS, Greenblatt DJ, et al. Therapeutic serum lidocaine and metabolite concentrations in patients undergoing electrophysiologic study after discontinuation of intravenous lidocaine infusion. Am Heart J. 1989;117(5):1060–4.
Jürgens G, Graudal NA, Kampmann JP. Therapeutic drug monitoring of antiarrhythmic drugs. Clin Pharmacokinet. 2003;42(7):647–64.
Burney RG, DiFazio CA, Peach MJ, Silvester MJ. Anti-arrhythmic effects of lidocaine metabolites. Am Heart J. 1974;88(6):765–9.
Gammaitoni AR, Alvarez NA, Galer BS. Pharmacokinetics and safety of continuously applied lidocaine patches 5%. Am J Health Syst Pharm. 2002;59(22):2215–20.
Gammaitoni AR, Davis MW. Pharmacokinetics and tolerability of Lidocaine patch 5% with extended dosing. Ann Pharmacother. 2002;36(2):236–40.
Campbell BJ, Rowbotham M, Davies PS, et al. Systemic absorption of topical lidocaine in normal volunteers, patients with post-herpetic neuralgia and patients with acute herpes zoster. J Pharm Sci. 2002;91(5):1343–50.
Aarons L. Population pharmacokinetics: theory and practice. Br J Clin Pharmacol. 1991;32(6):669–70.
Binder A, Bruxelle J, Rogers P, et al. Topical 5% lidocaine (lignocaine) medicated plaster treatment for post-herpetic neuralgia: results of a double-blind, placebo-controlled, multinational efficacy and safety trial. Clin Drug Investig. 2009;29(6):393–408.
Hans G, Sabatowski R, Binder A, et al. Efficacy and tolerability of a 5% lidocaine medicated plaster for the topical treatment of post-herpetic neuralgia: results of a long-term study. Curr Med Res Opin. 2009;25(5):1295–305.
Bryant MS, Simmons HF, Harrell RE, et al. 2,6-Dimethylaniline—hemoglobin adducts from lidocaine in humans. Carcinogenesis. 1994;15(10):2287–90.
Morris D, Podolski J, Kirsch A, et al. Population pharmacokinetics of telapristone (CDB-4124) and its active monodemethylated metabolite CDB-4453, with a mixture model for total clearance. AAPS J. 2011;13(4):665–73.
Huitema AD, Mathot RA, Tibben M, et al. Population pharmacokinetics of thioTEPA and its active metabolite TEPA in patients undergoing high-dose chemotherapy. Br J Clin Pharmacol. 2001;51(1):61–70.
Summary of product characteristics. UK SPC lidocaine 5% medicated plaster. Grünenthal Ltd, Uxbridge, Middlesex, UK, 13 Nov 2014.
Acknowledgements
Support for the pharmacokinetic analysis and interpretation of the data of the manuscript was provided by Michael Gautrois, department of Pharmacokinetics, Grünenthal GmbH, Zieglerstrasse 6, 52078 Aachen, Germany. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Roberta Bursi, Chiara Piana, and Irmgard Boesl are employees of Grünenthal. At the time of the analysis of the data, Dymphy Huntjens was an employee of Grünenthal. At the time of the analysis, Joachim Grevel was working under contract for EMF Consulting. This work was supported by Grünenthal GmbH, Zieglerstrasse 6, 52078 Aachen, Germany.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bursi, R., Piana, C., Grevel, J. et al. Evaluation of the Population Pharmacokinetic Properties of Lidocaine and its Metabolites After Long-Term Multiple Applications of a Lidocaine Plaster in Post-Herpetic Neuralgia Patients. Eur J Drug Metab Pharmacokinet 42, 801–814 (2017). https://doi.org/10.1007/s13318-017-0400-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-017-0400-7